

BY MATTHEW GU
1. Introduction
My first encounter with Holt-Oram Syndrome (HOS) occurred during a family holiday in China. In 2013, my parents’ friend gave birth to a baby boy named Jerry. His arrival was marked by joy but also great panic and concern. Jerry had been born with an atrial septal defect, a life-threatening condition where blood flowed abnormally through the wall between the upper chambers of his heart (British Heart Foundation). Fortunately, a top cardiovascular specialist performed a successful operation that saved Jerry’s life. At the time, I assumed this was simply an isolated case of a rare congenital heart defect. Little did I know, this experience would spark a much deeper interest years later.
Fast forward to 2017, when I encountered something strikingly similar. A 4-year-old girl who was a family friend came to the UK to have surgery on her right hand. She had a collection of unusual symptoms: speech difficulties, abnormalities in her thumbs, and—once again—an atrial septal defect. I remember being intrigued. The links between her symptoms seemed faint yet undeniably related. Was there a connection between the hands, the heart, and these developmental anomalies?
The peculiar nature of these conditions remained in the back of my mind until 2024, when I returned to China for another holiday. Jerry, now 11 years old, was as cheerful and friendly as ever. But this time, I noticed something unusual: a large scar along his right hand, extending from his thumb to his wrist. I quickly learned that he had experienced significant discomfort and difficulty moving his hand resulting in surgery.
It was at this moment that the pieces of the puzzle fell into place. The hand and the heart—two seemingly unrelated parts of the body—were somehow inextricably linked. I went home determined to understand the connection and that’s when I discovered the condition tying everything together: Holt-Oram Syndrome. Roughly 1 in 100,000 people have the condition (McDermott, Deborah A, 2015) and the probability of encountering two children with Holt-Oram Syndrome in such a short span of time is staggeringly low—approximately 0.0000000001% (JACC 2022), I was more likely to be struck by an asteroid. Yet here I was, having met Jerry and the young girl.
2. Holt-Oram Syndrome
Holt-Oram Syndrome (HOS), also referred to as “heart-hand syndrome,” is a rare genetic disorder caused by mutations in the TBX5 gene. The disorder is inherited in an autosomal dominant manner, meaning that only a single copy of the mutated gene inherited from one parent is sufficient enough to be expressed in the phenotype (Bajaj et al., 2017). There is also a 50% chance of passing the condition to offspring (Green and Cooper, 2019). HOS is named after Mary C. Holt and Samuel Oram who first reported the disease in 1960.
This condition primarily affects the development of the upper limbs and the heart, with individuals typically exhibiting congenital heart defects, such as atrial septal defects (ASD), as well as abnormalities in the thumbs and wrists (Briggs et al., 2018) which can range between mild and extremely severe. Jerry’s case was more mild than other cases found online. HOS can often be diagnosed through genetic testing and clinical examination (NORD, 2021) for example through echocardiography, where doctors will take an image of the heart to check for defects, or through electrocardiography (ECG) to monitor the electrical activities of the heart (Newbury-Ecob, 2017).
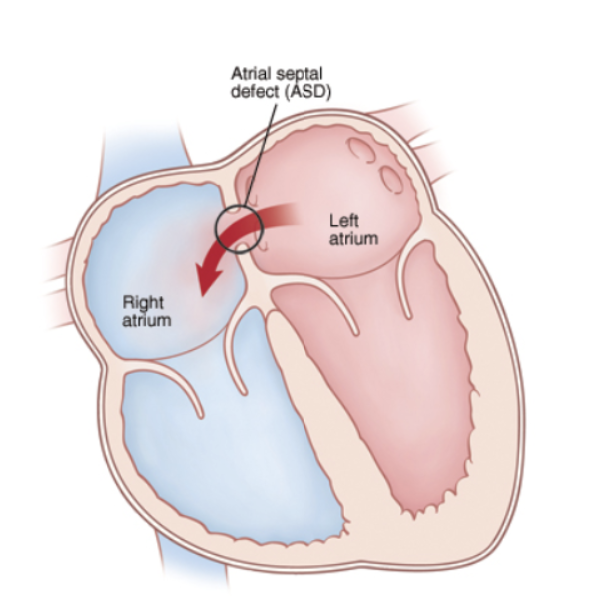
Figure 1: Diagram of the heart showing the movement of blood through the interatrial septum, causing Atrial septal defects.
All people with HOS have at least one abnormal wrist bone, which can often only be detected by X-ray (National Institute of Health, 2020). Most bone abnormalities include triphalangeal thumbs (TPT), a missing thumb, a thumb that looks like a finger, upper arm bones of unequal length or underdeveloped, partial or complete absence of bones in the forearm, and abnormalities in the collarbone or shoulder blade (MedlinePlus, 2020).
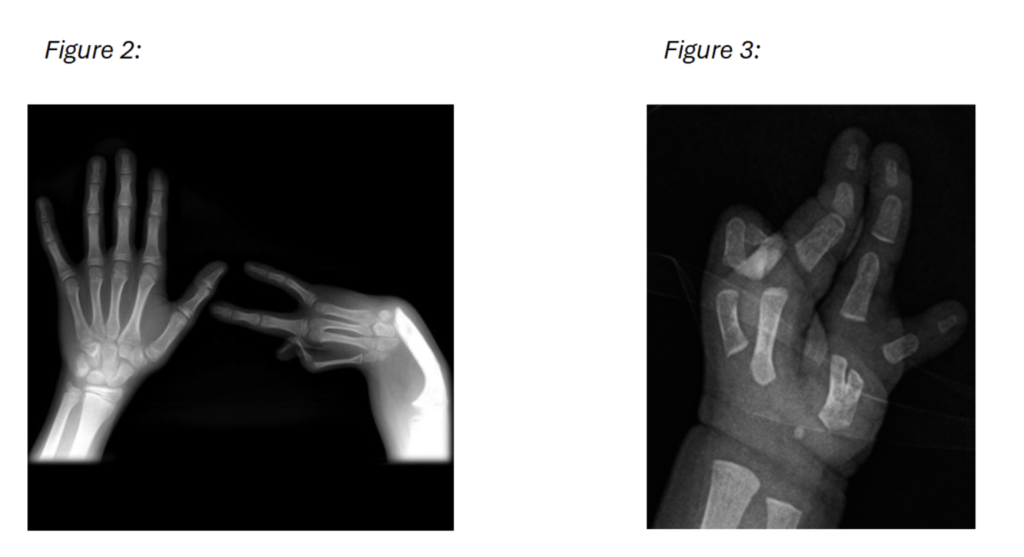
Figure 2 and 3: A frontal X-ray of the hands, complete absence of the radius and thumb on the right hand as well as additional hypoplasia of the 2nd metacarpal in figure 2.
About 75% of individuals with HOS also have congenital heart problems, with the most common being defects in the interatrial septum between the upper chambers of the heart (atrial septal defects) or the lower chambers of the heart (ventricular septal defect) (MedlinePlus, 2020). People with HOS may also have abnormalities in the electrical system that coordinates contractions of the heart chambers. Cardiac conduction disease can lead to bradycardia; rapid, ineffective contraction of the heart muscles (fibrillation); and heart block (MedlinePlus, 2020).
Other symptoms include early arthritis due to limited rotation of joints, abnormal bending of the 5th finger, and pigeon chest which affects the chest wall due to overgrowth of cartilage between the ribs and the sternum causing the chest to ‘stick out’
(Newbury-Ecob, 2017). Surprisingly, the lower limbs are not involved as embryogenesis in the 4-5th week occurs before lower limbs develop (Reddy, 2022).
3. Genetic Origin
Holt-Oram Syndrome (HOS) is caused by mutations in the TBX5 gene, located on the long arm of chromosome 12 (Patel, 2012). TBX5 belongs to a phylogenetically conserved family of genes characterised by a shared DNA-binding domain called the T-box. These genes play a critical role in development by encoding transcription factors—proteins that regulate the expression of other genes essential for key developmental processes.
The TBX5 gene specifically codes for T-box protein 5, a transcription factor integral to proper development (Jhang, 2015). This protein binds to DNA and acts as a regulatory switch, controlling which genes are activated or silenced during different stages of development. T-box protein 5 is particularly essential for the formation of the forelimbs and heart (Steimle, 2017). Mutations in TBX5 impair the function of T-box protein 5, preventing it from activating the correct genes at the appropriate times, which disrupts development. As a result, the consequences can be devastating and lead to significant abnormalities in affected individuals.
Structurally, T-box protein 5 is a monomeric protein made of a single polypeptide and does not form complex quaternary structures. In humans, the full-length protein consists of approximately 503 amino acids (NCBI Gene ID: 6914).
Moreover, T-box protein 5 also controls the expression of genes responsible for producing fibroblast growth factors (FGFs), particularly FGF-10 which is vital for limb bud development. FGFs are a family of cell-signalling proteins that are critical for cell growth. Disruption of FGF production or function due to TBX5 mutations can result in a wide range of developmental defects (Burgess, 1989). For example, failure of TBX5 to properly regulate FGF-10 can lead to Tetra-amelia syndrome, a rare recessive disorder characterised by the absence of one or both forelimbs (Niemann, 1993).
TBX5 plays a critical role as a tumour suppressor, helping to regulate cell growth and division by promoting apoptosis and maintaining cellular homeostasis (Bruneau, 2008). In the laboratory, TBX5 is also employed in reprogramming cocktails, a technique used to transform fibroblasts into cardiomyocytes (Zhao & Samaras, 2021). Impairment or mutation of TBX5 can increase the risk of cancer and abnormal tissue growth, as observed in HOS (Zhang, 2019).
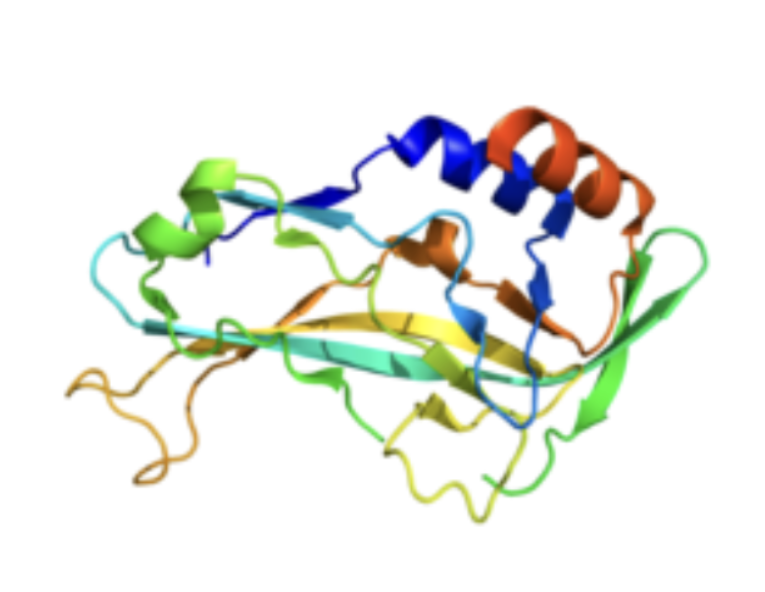
Figure 4: Tertiary structure of T-box protein 5 rendered by PyMOL (PDB 2X6U).
In a study conducted in 2001, researchers genetically modified mice to lack the TBX5 gene, which resulted in the embryos failing to survive gestation due to underdeveloped hearts. Mice with only one functional copy of the TBX5 gene exhibited significant structural defects, including enlarged hearts, atrial septal defects, and limb malformations, similar to those seen in HOS (Takeuchi JK, 2003).
4. Treatment
Firstly, atrial septal defects in individuals with HOS often require surgical intervention to close the holes in the heart (Smith et al., 2019). Additionally, cardiac conduction issues, such as bradycardia and heart block, may necessitate the implantation of a pacemaker to regulate heart function (Jones and Williams, 2017). Regular monitoring by a cardiologist is essential to ensure proper heart function throughout a patient’s life (Brown, 2018).
In addition to cardiac issues, limb malformations, including underdeveloped limbs or thumb abnormalities (White, 2020), can be addressed through surgical correction and physical therapy to improve functionality. Prosthetic devices are also commonly used to enhance mobility and daily functioning (Taylor, 2016). The Oxford Gait Laboratory carries out regular research on upper limbs and characteristics of hand use by assessing movement patterns to determine treatment plans. Surgical interventions often focus on reconstructing or improving the appearance and utility of affected limbs, particularly the hands (Miller et al., 2017).
Currently no medications are effective in treating the anatomical defects however antibiotic prophylaxis is often administered for patients suffering congenital heart problems (Basson, 2022).
Gene therapy represents a promising area of research aimed at correcting genetic mutations at their source. Advances in CRISPR-Cas9 technology enable researchers to target and potentially repair defective genes such as TBX5. CRISPR consists of a Cas9 protein, which acts like molecular scissors to cut the DNA, and guide RNA (gRNA), which directs the Cas9 protein to the specific location of the mutation. Scientists typically use viral vectors or lipid nanoparticles to deliver CRISPR components into the body. If the TBX5 mutation can be corrected early in development, it could prevent the heart and limb abnormalities associated with HOS (Doudna, 2016). Although this technology is still in its early stages, preclinical studies in animals have shown promising results.
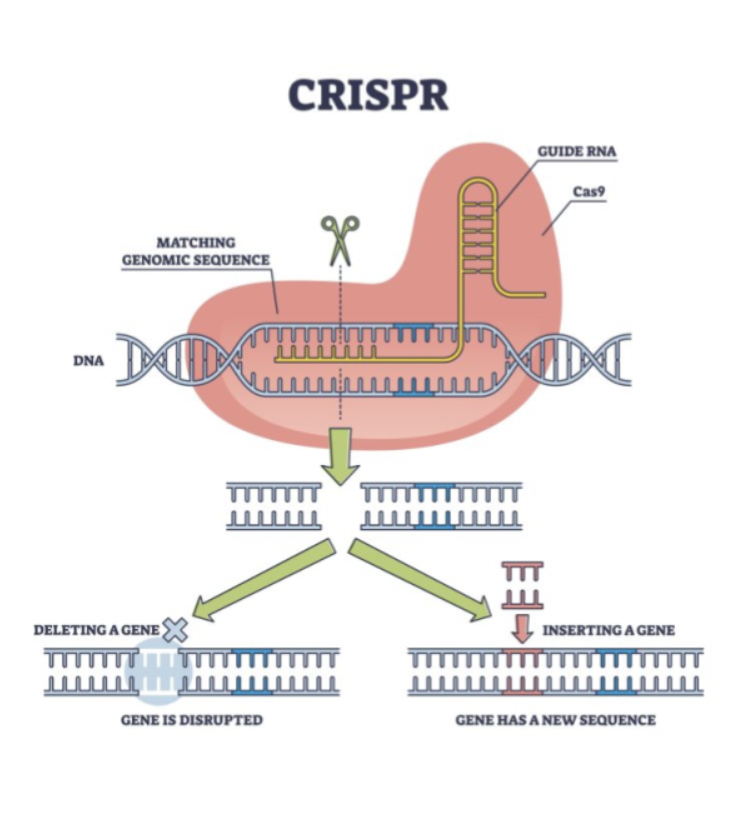
Figure 5: Labelled diagram outlining the structure and function of CRISPR technology.
Additionally, stem cell therapies are being explored, with a focus on using induced pluripotent stem cells (iPSCs) to regenerate damaged tissues in affected individuals. Efforts are also underway to develop improved diagnostic tools, such as genetic testing, for early detection of TBX5 mutations (Brown, 2019). Pharmacological treatments are being investigated as well, including targeting molecular pathways and proteins like FGF-10 (Burgess, 1989).
5. Rare Case
In 2021, an article by Professor M. Logan (KCL) detailed a large German family exhibiting an unusual overlap of Holt-Oram Syndrome (HOS) and Ulnar-Mammary Syndrome (UMS), which share similarities in bone deformities, but UMS also affects apocrine and mammary gland function (NORD, 2008). Across six generations, at least 17 family members were reported to carry duplications of both the TBX3 and TBX5 genes. Affected individuals experienced symptoms such as typical congenital heart defects and aortic stenosis (Logan, 2021). Of the 17 cases, 15 presented with cardiac malformations, with atrial septal defects (ASD) being the most common, affecting 5 individuals. One foetus exhibited a severe cardiac defect characterised by a hypoplastic left ventricle and a disproportion between the diameters of the aorta and pulmonary tracts. Duplication mutations of this type had never previously been observed.
6. Conclusion
In conclusion, I hope this article provides an insight into the fascinating nature of HOS, a condition that uniquely illustrates the connection between genetics and developmental abnormalities. While significant progress has been made in understanding HOS, ongoing research continues to offer hope for improved treatments and interventions, striving to alleviate the challenges faced by affected individuals.
7. Glossary
Aortic Stenosis: A condition where the aortic valve in the heart becomes narrowed, restricting blood flow from the heart to the rest of the body.
Apoptosis: The process of programmed cell death where cells are systematically dismantled and removed, crucial for normal development and tissue homeostasis.
Apocrine: A type of sweat gland that secretes a thicker, protein-rich fluid, often associated with body odour.
Autosomal Dominant: A pattern of inheritance where only one copy of a mutated gene is required to express a genetic disorder. Affected individuals have a 50% chance of passing the mutation to offspring.
Bradycardia: A slower-than-normal heart rate, usually fewer than 60 beats per minute, which may cause fatigue, dizziness, or fainting.
Cardiac Conduction Disease: A group of disorders affecting the heart’s electrical system, leading to problems with the heart’s rhythm and ability to pump blood effectively.
Cardiac Septal Defects: Abnormal openings in the septum (wall) that divides the chambers of the heart, including atrial septal defects (ASD) and ventricular septal defects (VSD).
Collarbone (Clavicle): The bone connecting the arm to the body, located between the sternum (breastbone) and the shoulder blade.
Congenital: A condition that is present from birth, which can be caused by genetic factors or environmental influences during pregnancy.
CRISPR-Cas9: A gene-editing tool used to precisely alter DNA in living organisms. It involves the Cas9 protein, which acts like molecular scissors, and guide RNA to direct it to specific DNA locations.
Electrocardiography (ECG or EKG): A medical test that records the electrical activity of the heart and is used to detect heart abnormalities.
Embryogenesis: The process by which an embryo forms and develops, beginning with fertilisation and continuing until the foetus is fully formed.
Fibroblast Growth Factor (FGF): A family of proteins that help regulate cell growth, development, and healing. FGF-10 is essential for limb development.
Fibrillation: A rapid, irregular, and ineffective heart rhythm, where the heart muscles contract erratically, often leading to poor blood circulation.
Gene Therapy: A technique that involves altering genes inside an individual’s cells to treat or prevent disease, sometimes by correcting genetic mutations.
Heart Block: A condition in which the electrical signals in the heart are delayed or blocked, causing the heart to beat too slowly or irregularly.
Hypoplasia: Underdevelopment or incomplete development of a tissue or organ, where it is smaller than expected.
Interatrial Septum: The wall that separates the left and right atria (upper chambers) of the heart.
Lipid Nanoparticles: Tiny particles made of lipids (fat molecules) used to deliver drugs or genetic material into cells, particularly in gene therapy or vaccine delivery.
Malformations: Abnormal formations or defects in body parts, typically due to issues during development.
Mammary Glands: Specialised organs in female mammals that produce milk to nourish offspring.
Metacarpal: The five bones in the hand that connect t
Monomeric Protein: A protein made of a single polypeptide chain that does not form complex structures with other proteins.
Mutations: Changes in the genetic code that can result in genetic disorders or variations in traits.
Phenotype: The observable characteristics or traits of an organism, such as appearance, behaviour, or physiological features, resulting from the interaction of its genetic makeup and environment.
Pluripotent: Describes cells that have the ability to develop into many different types of cells, particularly referring to stem cells that can give rise to all the cell types in the body.
Prosthetic Devices: Artificial tools or limbs that replace or enhance the function of missing or impaired body parts.
Quaternary Structure: The highest level of protein structure formed when multiple protein subunits combine to create a functional complex.
Sternum: The flat bone located in the chest, also known as the breastbone, which helps protect the heart and lungs.
Triphalangeal Thumbs (TPT): A condition where the thumb has an extra bone, making it appear as though the thumb has three phalanges (bones) instead of two.
Viral Vectors: Modified viruses used to deliver genetic material into cells in gene therapy, as viruses naturally have the ability to enter cells.
8. Bibliography
Bajaj, K., Saini, V., & Kapoor, A. (2017). Holt-Oram Syndrome: A Comprehensive Review. Indian Journal of Human Genetics, 23(3), pp. 306-312.
Basson, C. (2022). HOS treatment and management. theheart.org, Medscape Registration.Holt-Oram Syndrome.docx
Briggs, T., Bayliss, S., & Hudson, M. (2018). The Role of TBX5 in the Pathogenesis of Holt-Oram Syndrome. Journal of Medical Genetics, 55(5), pp. 271-278.
Brown, L. (2018). Cardiac care for congenital heart defects in Holt-Oram syndrome. Heart Health Journal, 25(3), pp. 22-30.
Bruneau, B. G. (2008). The developmental genetics of congenital heart disease. Nature, 451(7181), 943–948. https://doi.org/10.1038/nature06875
Burgess, W.H., & Maciag, T. (1989). The heparin-binding (fibroblast) growth factor family of proteins. Annual Review of Biochemistry, 58, pp. 575–606.
Green, P., & Cooper, M. (2019). Genetic counselling for autosomal dominant disorders: A guide for Holt-Oram syndrome. Journal of Genetic Medicine, 10(2), pp. 45-50.
Jhang, W.K., Lee, B.H., Kim, G.H., Lee, J.O., & Yoo, H.W. (2015). Clinical and molecular characterisation of Holt-Oram syndrome focusing on cardiac manifestations. Cardiology in the Young, 25(6), pp. 1093–1098.
Jones, R., & Williams, J. (2017). Managing cardiac conduction issues in Holt-Oram syndrome. Cardiology Today, 14(4), pp. 88-92.
Logan, 2021. TBX3 and TBX5 duplication: A family with an atypical overlapping Holt-Oram/ulnar-mammary syndrome phenotype.
Mackenzie, M.R., & Morris, A.R. (2018). The Clinical Manifestations and Diagnosis of Holt-Oram Syndrome. American Journal of Medical Genetics, 176(6), pp. 1124-1132.
McDermott, D.A., Fong, J.C., & Basson, C.T. (2015). Holt-Oram Syndrome. In: Adam, M.P., Ardinger, H.H., Pagon, R.A., et al. (eds.), Gene Reviews. University of Washington, Seattle. [Online] Available at: https://www.ncbi.nlm.nih.gov/books/NBK1460/
Miller, T., Davis, J., & Evans, S. (2017). Orthopaedic interventions in limb malformations caused by Holt-Oram syndrome. Clinical Orthopaedics Review, 32(6), pp. 35-41.
National Institute of Health (NIH). (2020). Holt-Oram Syndrome. Genetic and Rare Diseases Information Centre (GARD). Available at: https://rarediseases.info.nih.gov/diseases/6767/holt-oram-syndrome
National Organization for Rare Disorders (NORD). (2021). Holt-Oram Syndrome. NORD. Available at: https://rarediseases.org/rare-diseases/holt-oram-syndrome/
NORD (2008). Schinzel syndrome.
Parker, J. (2015). A multidisciplinary approach to treating Holt-Oram syndrome. Medical Advances, 21(1), pp. 12-19.
Patel, C., Silcock, L., McMullan, D., Brueton, L., & Cox, H. (2012). TBX5 intragenic duplication: a family with an atypical Holt-Oram syndrome phenotype. Journal of Medical Genetics, 49(8), pp. 501-507.
Rasagnya M Reddy (2022). NIH: A rare Variant and Unusual Presentation of HOS in a Child.
Ruth Newbury-Ecob. Holt-Oram Syndrome Diagnosis. Contact.org.uk.
Smith, G., Roberts, D., & Clark, A. (2019). Surgical treatment of congenital heart defects in Holt-Oram syndrome. Journal of Paediatric Surgery, 18(2), pp. 58-63.
Takeuchi, J.K., Ohgi, M., Koshiba-Takeuchi, K., Shiratori, H., Sakaki, I., Ogura, K., et al. (2003). Tbx5 specifies the left/right ventricles and ventricular septum position during cardiogenesis. Development, 130(24), pp. 5953–5964.
Taylor, E. (2016). Physical therapy for children with limb abnormalities due to Holt-Oram syndrome. Rehabilitation Review, 12(3), pp. 18-23.
White, R. (2020). Understanding limb malformations in Holt-Oram syndrome. Paediatric Orthopaedics Journal, 40(7), pp. 102-110.
Zhang, M., Lin, L., & He, Q. (2019). The Role of TBX5 in Cancer Biology and Beyond: A Review. Frontiers in Cell and Developmental Biology, 7, 289. https://doi.org/10.3389/fcell.2019.00289
Zhao, C., & Samaras, S. E. (2021). Reprogramming Fibroblasts into Cardiomyocytes: The Role of TBX5 and Other Key Factors. Journal of Molecular Biology, 433(17), 166998. https://doi.org/10.1016/j.jmb.2021.166998