

BY AUSTIN LAM
BDNF in Depression Lowered BDNF levels have been associated with an increased frequency of depressive symptoms, summarising the neurotrophic hypothesis. Postmortem studies exhibit lowered levels of BDNF coupled with its receptor TrkB[31], with emphasis of reduced BDNF mRNA in regions such as the hippocampus[32] and amygdala[33]. Moreover, those who died of complete suicide have been found to have reduced BDNF compared to controls[34]. Though depressed patients exhibit lower circulating BDNF levels, such levels can be ameliorated and even standardised through antidepressant treatment35, an increase shared amongst animal models equally[36]. Animal models of depression have also revealed antidepressant-like results through BDNF, as a plethora of antidepressants increase BDNF expression which could in turn aid in its action[37]. Reliance on chronic prescription may point towards a type of correlation with chronic alteration in neurotrophic function.
Methylation of the BDNF promotor has also remained higher in depressed patients with implications of suicidal ideation, whilst its elevation at promoter IV has relations to depressive symptoms[38]. The same case can be made from brain samples of those who have died by suicide, confirmed to be a gene specific increase in CpG methylation considering an unaffected global DNA methylation level[39]. Associations between suicidality and BDNF hypermethylation prove DNA methylation to be impactful in terms of [40]BDNF expression regulation.
The genetic relevance of single nucleotide polymorphisms (SNPs) cannot go ignored in the context of depression. One of the most extensively studied is the Val66Met (rs6265) SNP, occurring at nucleotide 196 leading to a substitution of valine to methionine at codon 66 in a region of the BDNF gene encoding the pro-domain. The polymorphism disrupts activitydependent secretion of BDNF and intracellular trafficking[41], hence reflective of less BDNF activity. Suggestions of this SNP’s ability as a predictor of neuropsychiatric disorder susceptibility have been investigated, but various stances have surfaced according to its role in depression. Some studies stated that the Met allele increased depression vulnerability[42] , strengthened by meta- analysis specifically in male Met-carriers[43] and have been associated with suicidal behaviour most notably in depressed patients[44]. Contrastingly, this view has also been denied, with findings of no statistically worthy association between val66met, or rather any genetic variants with depression[45,46]. A variety of SNPs exist in the BDNF gene in humans altering function and human phenotype expression.
Contradictory findings accentuate the importance of gene-environment interaction especially when the consequence of the SNP can be inconsistent under equal conditions. Shkundin & Halaris (2023) demonstrated this, where Met-carriers exhibited higher depressive symptoms in the presence of low levels of neglect as opposed to higher neglect increasing solely in the Val/Val genotype[47]. A meta-analysis conducted with a total of 21060 participants proved the Met allele in the BDNF Val66Met polymorphism to moderate the relationship between depression and stress exposure[48]. Evidence of the polymorphism modulating effects of factors such as chronic stress or early life adversity on depression frames the val66met SNP to be insufficient to be a direct cause of depression, but rather a risk factor that engages sensitively with environmental stressors influencing their susceptibility.
BDNF levels are often high in human serum, with approximately a 100- to 200- fold difference when compared with serum levels as BDNF is released during the clotting process[49]. Over 90% of blood BDNF is stored in platelets[50], with its importance raised through abnormally low serum and plasma BDNF levels in depressed patients[51] . Karege et al[52] initially proposed a correlation between serum and brain BDNF, meaning peripheral measurements of BDNF would prove to be a sufficient biomarker. A cautious approach must be taken however, as the relationship between CNS and platelet BDNF requires further research, supposedly incapable of passing through the BBB. In patients with depression, unaltered whole blood BDNF levels as opposed to lower serum and plasma BDNF[53] point towards the release mechanism of BDNF at fault, likely platelet dysfunction[54]. Differentiating between healthy controls and depressed patients lies in platelet release of BDNF upon stimulation.
In murine models, the forced swim test and learned helplessness model of despair exhibit antidepressant-like outcomes after BDNF infusion into the hippocampus and midbrain[55]. Depressive tendencies evolved from heterozygous mice in terms of the BDNF allele, and genetic uniqueness which cause differences in BDNF signaling remain a promising explanation. Regardless, it would be wise to take into account that sensitivity and reliance may produce inconsistent results[56]. It is important to note that unlike humans, BDNF is not contained in mice platelets, as the BDNF gene is not transcribed to a sufficient level, such that it is undetectable in mouse megakaryocytes.
Central and peripheral inflammation
Armed with the knowledge that the pathophysiology of depression has many factors, it does not arise by BDNF dysregulation alone. The contribution of inflammation has been elucidated, as the etiology of depression may well be associated with the severity of the immune response. Neither factor functions independently when it comes to inducing depressive behavior. Inflammation occurs as the immune system is activated, triggered by noxious stimuli as an adaptive response, aiming to remove such injurious stimuli often contributing to the healing process. As a medical term with rich history, the first four cardinal signs characterised by redness (rubor), swelling (tumor), heat (calor) and pain (dolor) were initially documented in ancient Rome by Celsus before the fifth, namely loss of function (functio laesa), by Galen[57]. The main aspect of classical inflammation mentions a localised inflammatory focus which aims to isolate and eliminate the damage factor thus allowing for subsequent repair. A controlled inflammatory response is a favorable second-line defense when exposed to infectious agents but turns destructive when dysregulated. Though traditionally viewed as a type of protective reaction, not all states of inflammation have a definite physiological counterpart, exemplified through conditions such as gout or obesity. Apart from infection-induced inflammation, other versions of inflammatory responses are only understood under a pathological context rather than an adaptive state, challenging the normalized perspective on inflammation. Regardless, evolution has most likely tailored inflammation towards an attempt at restoring homeostasis. Upon tissue injury, harmful stimuli trigger the acute inflammatory response with a chemical signalling cascade leading to chemotaxis of circulating leukocytes which produce inflammatory cytokines upon activation, delivering blood components from vascular responses to the site of injury[58]. Failure to resolve inflammation propagates noxious inflammatory stimuli rendering it chronic, involving infiltration of macrophages and lymphocytes which are primary inflammatory cells replacing neutrophil infiltrate. Further failure and persistence of inflammatory stimulus led to granuloma formation, an aggregation of immune cells as a last attempt to ‘wall off‘ an agent[59] . Chronic inflammation is a key contributor in many diseases ranging from cardiovascular diseases, cancer, diabetes, rheumatoid arthritis, allergic asthma, chronic obstructive pulmonary disease (COPD), to Alzheimer’s and Inflammatory Bowel Disease (IBD). Risk factors consist of age, obesity, diet, stress and diet, some of which often occur alongside depression.
Inflammation heavily depends on leukocyte recruitment, and the mechanisms of leukocyte trafficking has been investigated from inflammation which has been induced by microbial pathogens, or rather their evolutionary conserved molecular patterns[60]. Signals derived from damaged tissues or pathogens are recognized by pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs) which are expressed as surface receptors on immune cells therefore mediating an inflammatory response. PRRs are activated and bind to conserved microbial products, a class of microbial inducer, such as lipopolysaccharides (LPS) from bacterial cell walls known as pathogen-associated molecular patterns (PAMPs)[61]. In contrast, inflammation is not necessarily purely caused by infectious agents, and damage-associated molecular patterns (DAMPs), are released during tissue or cell damage. It is associated with host-related injury with canonical connotations to damage which gives it its name. This process is known as ’sterile inflammation’ which happens in the absence of pathogens and their products, meaning etiologies of inflammation can be both infectious and non-infectious. Conserved molecular patter recognition by PRRs respond by initiating intracellular signaling cascades vital for the inflammatory response, suppressing anti-inflammatory gene expression. Activation of PRRs commonly activates the canonical NF-κB pathway through pro-inflammatory mediator production, regulating the transcriptional induction of pro-inflammatory cytokines, chemokines, and additional inflammatory mediators in different types of innate immune cells These cellular processes and their respective engagement with Toll-like receptors (TLRs) and Nod-like receptors (NLRs) results in the nuclear translocation of transcription factors such as NF-κB and AP-1, leading to pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6, with downstream induction of reactive oxygen species[62]. TLR-stimulated pro-inflammatory cytokine is enhanced by ROS production with a positive feedback loop, as an excess ROS maintains chronic inflammation.
Immunity can be classified as ‘innate’ or ‘adaptive’, where innate is the first line of defense due to innate myeloid cells and lymphoid cells offering a fast-acting response being in proximity, maintaining barrier sites. Part of inflammation triggers the adaptive immune response from dendritic cell antigen presentation to those of the adaptive immune system, also termed antigen-presenting cells (APCs). Adaptive immune cells are based on antigen specific receptors, able to perform clonal expansion upon activation by an antigen[63]. The adaptive immune system comprises of cells such as antigen-specific T cells which proliferate in adequate conditions, and B cells which produce antibodies by differentiating into plasma cells.
Inflammation also triggers a response from the whole body, which includes influence on the central nervous system (CNS). Multiple lines of evidence point towards bidirectional communication between the immune and central nervous system (CNS)[64], essential in regulation of synaptic plasticity and transmission. Such crosstalk is driven through the bloodbrain barrier, which has both a physical and metabolic barrier restricting pathogens as well as solute diffusion into the blood. Its composition of endothelial cells (ECs), pericytes (PCs), capillary basement membrane along with astrocyte end-feet prevents toxic insults from affecting the brain[65]. Consisting of endothelial cells connected by tight junctions, CNS homeostasis is effectively maintained by regulation of circulating substances therefore providing an optimum microenvironment. Chronic inflammation is able to compromise the integrity of the BBB making infiltration of immune cells into the brain possible. This is further aggravated by oxidative stress from reactive oxygen species (ROS) due to activation of microglia and astrocytes as well as inflammatory cytokines. Eventually, this gives the opportunity for immune mediators to enter the brain parenchyma exerting neuroinflammation. Prior to a ’second strike’, chronic inflammation primes a weakened BBB, making it more vulnerable to future inflammatory insults. Acute inflammatory responses observed from surgery, infection or trauma first upregulate peripheral cytokines, DAMPs and PAMPs function as a secondary trigger. Such an event would entail risk of different magnitudes of neuronal damage and synaptic dysfunction[66], possibly explaining the prevalence of perioperative neurocognitive disorders such as delirium which result in long-term poor outcomes. It is important to note that in older, chronically ill patients, systemic inflammation leads to an inflammatory response on a larger scale leading to neurodegeneration. Research has also discovered that inflammation and pro-inflammatory cytokine levels can be regulated through the ’inflammatory reflex’, where signalling of the afferent vagus nerve is functionally related with the efferent vagus nerve-mediated output mechanism[67] .
Microglia are resident immune cells in the CNS with a function comparable to guards, screening and sensing nearby inflammatory cues, seated within the brain parenchyma. The versatility of their plasticity allows them to effectively alter their cell morphology in response to such signals. In normal state, a ramified morphology of a small soma is maintained. However, in circumstances where microglial activation may take place such as from peripheral inflammation by systemic lipopolysaccharide (LPS) administration, microglia exhibit a much more aggressive pro-inflammatory phenotype, proved by galectin-3-positive microglia, a disease-associated phenotypic marker for microglia[68]. Immunologically activated microglia can be characterized by an amoeboid morphology, increasing cytokine and ROS production. The function of microglia can equally be affected by the essential amino acid tryptophan mostly metabolized through the kynurenine pathway, where an increase in tryptophan metabolism can be caused by either chronic stress or inflammation. When the rate-limiting enzyme indoleamine-2,3-dioxygenase (IDO) is upregulated, it aids in conversion of tryptophan into kynurenine in microglia upon activation, and several cytokines can upregulate IDO, an event that exists both in the brain or in the periphery. The kynurenine pathway has two main branches which ultimately result in generation of 3-hydroxykynurenine (3-HK), quinolinic acid (QA) via activation of kynurenine 3-monooxygenase (KMO), or conversion into kynurenic acid (KA) (fig.2). 3-HK is a toxic metabolite which can induce oxidative damage, with neuronal damage mediated by free radicals. The neurotoxic effects of the NMDA receptor agonist QA could potentially be modulated by KA, as an antagonist of NMDA receptors with opposing neuroprotective effects. QA is a byproduct of the pathway, produced in microglia, and 3-HK acts as a precursor due to its ability to pass the BBB along with kynurenine and tryptophan. It should be logical to suggest that metabolite imbalance may well contribute to plenty of abnormal behavior relevant to neuropsychiatric diseases.
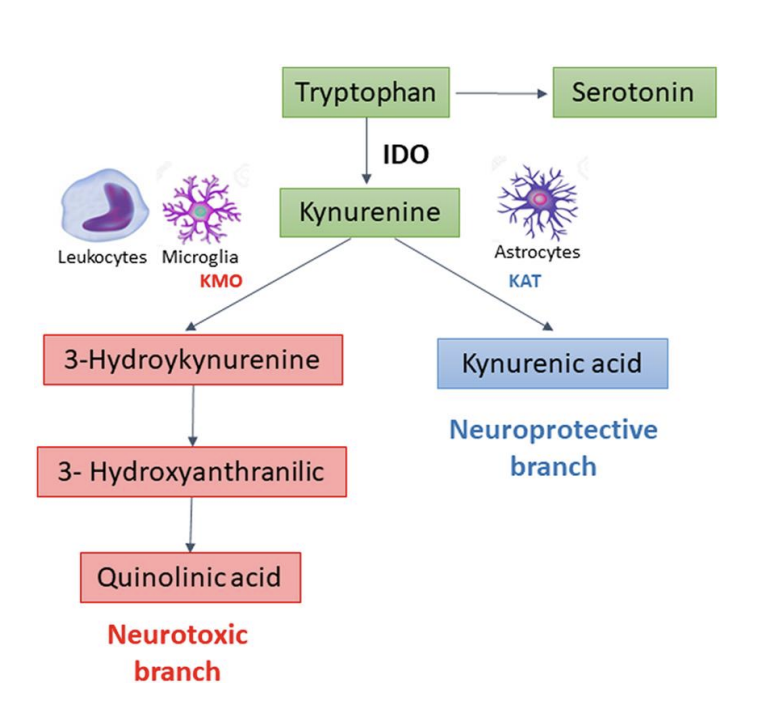
Figure 2 – The kynurenine pathway (KP) of tryptophan metabolism. Image source: Colpo, G.D., Venna, V.R., McCullough, L.D. and Teixeira, A.L. (2019). Systematic Review on the Involvement of the Kynurenine Pathway in Stroke: Pre-clinical and Clinical Evidence. Frontiers in Neurology, 10. doi:https://doi.org/10.3389/fneur.2019.00778.